

El Síndrome Cornelia de Lange (SCdL), es una enfermedad congénita, con compromiso multisistémico, compuesta por una serie de síntomas y características clínicas diversas, discapacidad intelectual, acortamiento de las extremidades, y retardo en el crecimiento. Básicamente, se trata de un desorden que afecta física, cognitiva y conductualmente al niño. Se estima una prevalencia entre 1 en 10.000 a 30.000 en nacimientos vivos, lo cual no puede ser despreciable. Las investigaciones a nivel molecular, revelan que la etiología del SCdL, se debe a variantes genéticas de los componentes regulatorios o estructurales del complejo de las cohesinas. Esto es particularmente importante, ya que se ha identificado que las cohesinas tienen la función de establecer la cohesión de las cromátidas hermanas, y se ha observado que desempeñan un papel importante en la expresión de genes, y en la organización 3D del genoma. Las mutaciones en el gen NIPBL, representa la causa de aproximadamente el 70% de los casos; sin embargo, entre el 5%-10%, de los niños con SCdL, se asocian a mutaciones en los genes SMC1A, SMC3, RAD21, HDAC8, BRD4 y ANKRD11 genes (1)(2)(3)(4)
Las características fenotípicas clásicas, se aprecian en la figura 1, aunque no siempre se presentarán de la misma forma, todo depende de las mutaciones presentes, y del compromiso que representen. En general, además de las características oculares, el surco labial se afecta, las orejas se implantan más bajas, y la nariz se ve más corta y respingada. (2)

Figura 1. Caracterización fenotípica del SCdL. a. Presentación clásica en niño con variación en gen NIPBL. b. Presentación no clásica en niño con variación en gen NIPBL. c. Adulto con variación en gen NIPBL. d. Presentación no clásica con variación en gen SMC1A. e. Fenotipo clásico en variante SMC3. f. Fenotipo no clásico en niño con variante RAD21 . g. Fenotipo no clásico en niño con variante HDAC8. h. Presentación no clásica en niña con variante ANKRD11 (2)
Manifestaciones oculares
Los signos clínicos oculares dependerán de la variedad mutagénica que se presente, se han identificado como los más comunes: tricomegalia, sinofridia, hirsutismo de las cejas, pigmento en el anillo peripapilar y miopía. En menor frecuencia, aproximadamente en 5% de los casos se observa hipermetropía, ptosis, blefaritis, hendidura palpebral pequeña, telecanto, fisuras palpebrales direccionadas inferiormente, microcórnea leve, estrabismo (especialmente endotropias), nistagmus, y anormalidades del nervio óptico. En un porcentaje alto (67%-80%) se ha encontrado obstrucción del conducto nasolagrimal uni o bilateral. La heterogeneidad de los hallazgos no termina ahí, también se ha reportado la presencia de macroftalmia, catarata y glaucoma, aunque no son tan prevalentes. Aun así, los casos más raros manifiestan corectopia, membrana pupilar parcial y adelgazamiento del iris. Ver figura 2. (2)(5)
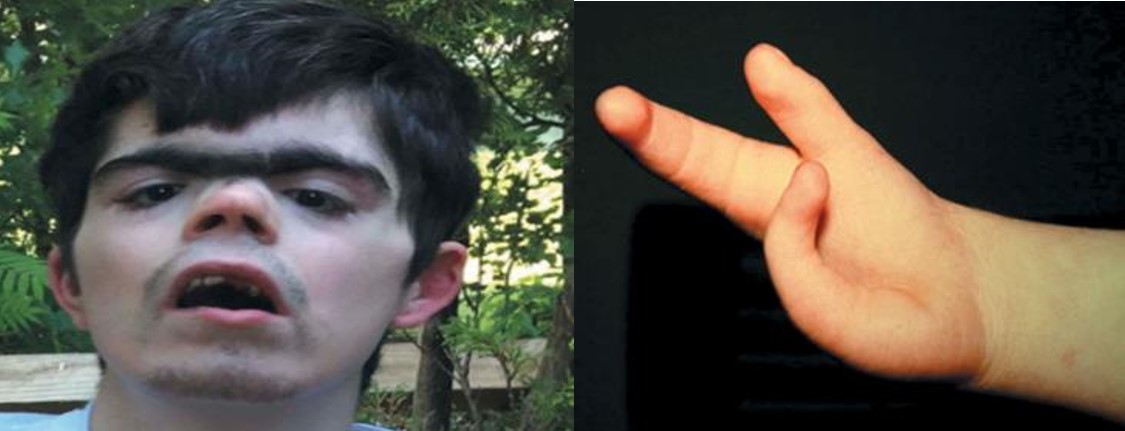
Figura 2. Se observa el hirsutismo de cejas, sinofridia, telecanto, tricomegalia, nariz respingada y corta, labio superior en forma de “v” invertida, surco naso labial liso y largo, fisuras palpebrales inclinadas inferiormente. A la derecha se aprecia la reducción de extremidad superior. (5)
El segmento posterior también es escenario de una variedad de hallazgos además del anillo peripapilar, se puede ver: disposición temporal de los vasos maculares, palidez retiniana, hipoplasia del nervio óptico, nervio óptico inclinado, y cambios pigmentarios retinales.(5)

Conclusión
El SCdL requiere manejo interdisciplinario, no solo en Oftalmología y Optometría en el manejo del estado refractivo, las alteraciones potenciales de la superficie ocular, el riesgo de ambliopía, el drenaje del sistema lagrimal, etc, sino también derivar al paciente a servicios como: psicología, medicina interna, ortopedia, terapia de lenguaje, neurólogo, gastroenterólogo, etc.
Referencias:
1. Latorre-Pellicer A, Ascaso Á, Trujillano L, Gil-Salvador M, Arnedo M, Lucia-Campos C, et al. Evaluating face2gene as a tool to identify cornelia de lange syndrome by facial phenotypes. Int J Mol Sci. 2020;21(3).
2. Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet [Internet]. 2018;19(10):649–66. Available from: http://dx.doi.org/10.1038/s41576-018-0031-0
3. Sarogni P, Pallotta MM, Musio A. Cornelia de Lange syndrome: From molecular diagnosis to therapeutic approach. J Med Genet. 2020;57(5):289–95.
4. Avagliano L, Parenti I, Grazioli P, Di Fede E, Parodi C, Mariani M, et al. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin Genet. 2020;97(1):3–11.
5. Shi A, Levin A V. Ophthalmologic findings in the Cornelia de Lange syndrome. Ophthalmic Genet [Internet]. 2019;40(1):1–6. Available from: https://doi.org/10.1080/13816810.2019.1571617



{kind=link}