

La amaurosis congénita de Leber (ACL) es una distrofia retiniana de aparición temprana; por lo tanto, será clave su detección en los primeros años de vida, especialmente antes de cumplir el primer año. A esta patología también se le ha conocido como retinitis pigmentosa congénita. El problema real con esta entidad, es que en un principio el fondo de ojo puede tener apariencia normal, y, en un corto tiempo, se empiezan a evidenciar los cambios pigmentarios, las alteraciones en el desarrollo y el nistagmus errático. Entre las manifestaciones clínicas más relevantes, se ha descrito que la agudeza visual puede oscilar entre 20/400 y no percepción de luz, lo cual la cataloga entre el espectro de distrofias hereditarias más graves. En cuanto a la parte refractiva, suelen estar asociadas a hipermetropías > 5 D. los niños también pueden presentar ausencia de reflejo pupilar y dependiendo de la mutación genética, también pueden presentar queratocono. Entre otras alteraciones, se puede mencionar el constante frote de los ojos (signo de Franceschetti), y respuestas electrorretinográficas casi indetectables. Las manifestaciones clínicas pueden variar de manera numerosa debido al tipo de mutación que tenga el niño. Por esta razón, algunos infantes tendrán retraso en el desarrollo cognitivo, mientras que otros manifestarán una inteligencia normal.1
Considerando ahora aspectos más específicos, como se mencionó anteriormente, la valoración de la retina puede ser normal o presentar signos muy leves en fases tempranas de la patología; sin embargo, la aparición de un moteado en el EPR, y adelgazamiento vascular, pueden ser signos tempranos de alarma. Posteriormente, se podrá evidenciar la presencia de retinopatía tipo sal y pimienta, palidez del nervio óptico, y la acumulación de pigmento en retina tipo espícula ósea y numular. Ahora bien, debido a la heterogenicidad semiológica de la ACL, se hace indispensable el estudio genético, ya que se han desarrollado estrategias de tratamiento según el genotipo específico de cada manifestación; y de ello, también dependerá el pronóstico, y el manejo multidisciplinar de condiciones sistémicas asociadas. Ver Figura 1.2
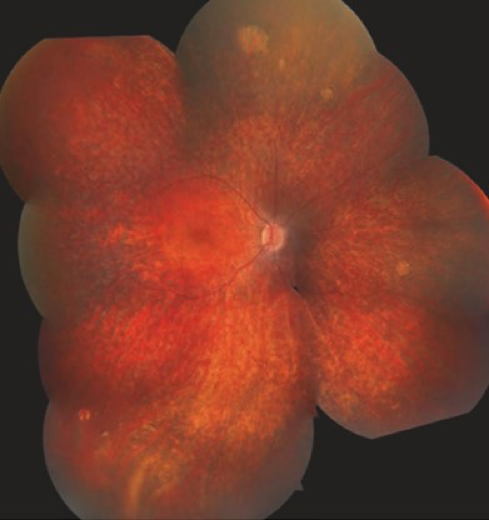
Es necesario destacar que la ACL puede ser una entidad clínica aislada o se puede integrarse como parte de síndromes como Senior Loken o Joubert. Es una de las distrofias que mayor componente genético tiene, y se ha dicho que está causada por variantes en por lo menos 25 genes como: RPE65, GUCY2D, NMNAT1, CEP290, AIPL1, y RDH12. De lo anterior se desprende que se han identificado características clínicas específicas y diferenciadoras según el genotipo, como por ejemplo la atrofia macular temprana atribuida a las mutaciones en RDH12, CRX, CRB1 y NMNAT1. Otro ejemplo, es el papel del gen AIPL1, que se localiza principalmente en los segmentos externos de los bastones, el cual reduce la concentración de guanosín monofosfato cíclico (cGMP). Continuando, el gen CRB1 tiene otro papel importante, ya que se localiza en la membrana de las células de Müller y tiene una función estructural proteica. y si siguiéramos, no podríamos dejar de nombrar a los genes LRAT y RPE65, que se localizan en el EPR, y están involucrados en el ciclo visual. Con base en esto, las mutaciones en estos genes importantes derivan en daño de la estructura y funcionamiento de la retina. 2,3
Dado lo complejo de la patología, se asevera que el tratamiento también lo es; de hecho, la primera instancia está dada para el alivio de complicaciones relacionadas como la catarata, anormalidades de la interfase vitrorretiniana, queratocono, y edema macular cistoide. Por otro lado, se ha descrito la recomendación de una dieta balanceada con propiedades antioxidantes rica en frutas y vegetales. Otros hábitos serán adoptados, como la protección contra los rayos UV, y el no fumar para evitar el estímulo de daño retinal por estrés. En instancias más tecnológicas, los esfuerzos en la investigación actual están enfocados en mejores procesos de desarrollo de terapia génica, dadas las bases monogénicas del ojo y su inmunoprivilegio. Por esto se está hablando de suplementación de genes, que consiste en el suministro de ADN al núcleo celular a través de un vector viral. Este proceso promueve la correcta transcripción de una proteína funcional que en condiciones de ACL falla por la mutación del gen original; de esta manera, la funcionalidad proteica se recupera, y a su vez el metabolismo retiniano. 2,3,4
Otros objetivos de investigación, está en el desarrollo de moduladores del ciclo visual, en términos de poder reducir la acumulación de derivados metabólicos, o la administración de componentes faltantes. Por ejemplo, se ha identificado que en algunas mutaciones que conducen a ACL, el todo- trans- retinal no se puede reconvertir en 11- cis- retinal, lo que conduce a degeneración de la estructura retinal y pérdida irreversible de la visión. De tal manera que, si se pudiera suplementar desde una fuente exógena el 11- cis- retinal, se podría prevenir la degeneración de los fotorreceptores. Tanto se ha luchado, que también se está trabajando en dispositivos optoelectrónicos, cuya finalidad es transformar una señal óptica en eléctrica y viceversa, para compensar la funcionalidad perdida de la retina, restaurando el curso normal de transformación del estímulo luminoso.2,4
El profesional de la salud visual debe recordar la importancia de detectar tempranamente signos de ACL, promover el manejo interdisciplinario donde el genetista esté ampliamente involucrado, y formar parte de la rehabilitación del paciente, donde los servicios de Baja Visión serán una herramienta indispensable en el apoyo al niño y al futuro adulto.
Referencias
1. Tsang SH, Sharma T. Leber congenital amaurosis. Adv Exp Med Biol. 2018;1085:131–7.
2. Varela MD, De Guimaraes TAC, Georgiou M, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Current management and clinical trials. Br J Ophthalmol. 2021;106(4):445–51.
3. Huang C-H, Yang C-M, Yang C-H, Hou Y-C, Chen T-C. Leber’s Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes (Basel). 2021;12(1261):2–18.
4. Chiu W, Lin TY, Chang YC, Lai HIAM, Lin SC, Ma C, et al. An update on gene therapy for inherited retinal dystrophy: Experience in leber congenital amaurosis clinical trials. Int J Mol Sci. 2021;22(9).



{kind=link}